Yes, if the device continues to design and intended for the specific use that is within the regulation you say applies. If your promotional material or labeling exceed what is described, you are likely no longer exempt.
The FDA states:
510(k) Exemption
When a generic type of product (often identified by its product code within a classification regulation) becomes exempt from the 510(k) requirements, future products of that generic type will not be required to obtain 510(k) clearance prior to marketing – but, the exemption is subject to the .9 limitations. Each classification regulation part (21 C.F.R. Parts 862 – 892) includes a .9 section (e.g., 21 C.F.R. § 862.9). This section is often overlooked for the more substantive, device-specific sections later in the Part. However, the .9 limitation is crucial in understanding what is and is not 510(k)-exempt.
The .9 limitation says that a device of the generic type in a 510(k)-exempt classification regulation is exempt so long as its characteristics were “existing and reasonably foreseeable” at the time the generic type of device became exempt from the 510(k) requirements. The .9 regulation elaborates on what this phrase means by way of examples. Specifically, a 510(k) is required if the proposed device:
-
has a “different” intended use as compared to the generic device type; or
-
uses a different “fundamental scientific technology” compared to the generic device type.
Here is an example:
|
510(k) Exempt
|
 |
|
510(k) Required
|
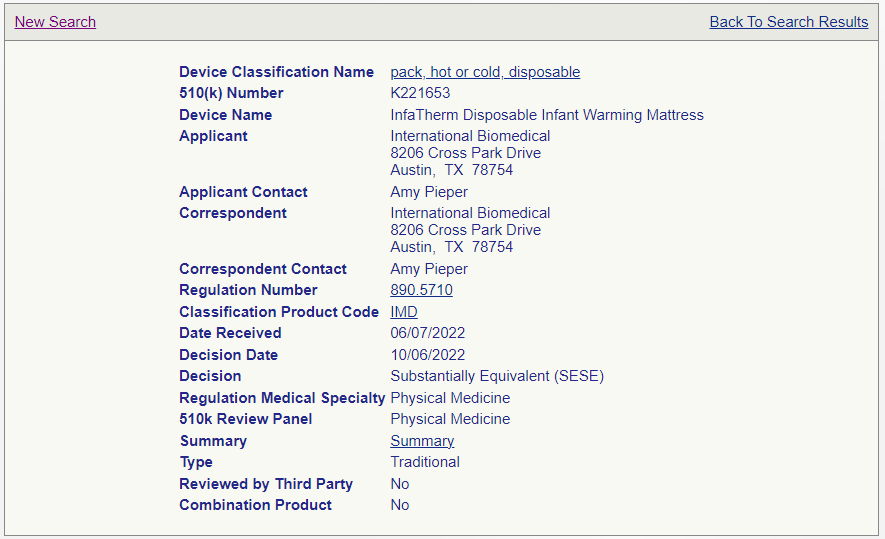  |


Certain claims, indications for use, materials, or sterility could impact whether you can still state your device is Exempt from 510(k).
Additional examples:
| 880.5090 | Liquid Bandage. Exemption is limited to uses as a skin protectant. | |
| 878.3250 | External Facial Fracture Fixation Appliance. 510(k) exempt only if the device is made of the same materials that were used in the device before May 28, 1976 |
|
| 876.5160 | Urological Clamp. Exemption does not include devices for internal use. | |
So what’s the risk?
- Receive a FDA letter that requires action (483, Warning Letter, Untitled Letter).
- Required to cease promotion & distribution.
- May be required to recall product and send notices to distributors and customers.
What’s the cost?
- Time
- Money
- Negative visibility (publicity)
- History of non-compliance now known to FDA
If you want an assessment of your currently marketed products and promotion, please reach out for an assessment of adequacy of compliance and claims substantiation.
